For many years, ribosome research in mammals has lagged far behind that of budding yeast. This is largely because yeast genetics (e.g. synthetic lethality and suppressors screens), and later yeast pre-ribosome purification by tandem affinity chromatography, proved extremely powerful for the identification of the >200 ribosome synthesis trans-acting factors identified to date in this experimental model [1]. This is probably also a reflection of the largely acknowledged assumption, mostly based on sequence conservation of putative human homologs of yeast assembly factors, that ribosome synthesis is largely conserved throughout eukaryotes.
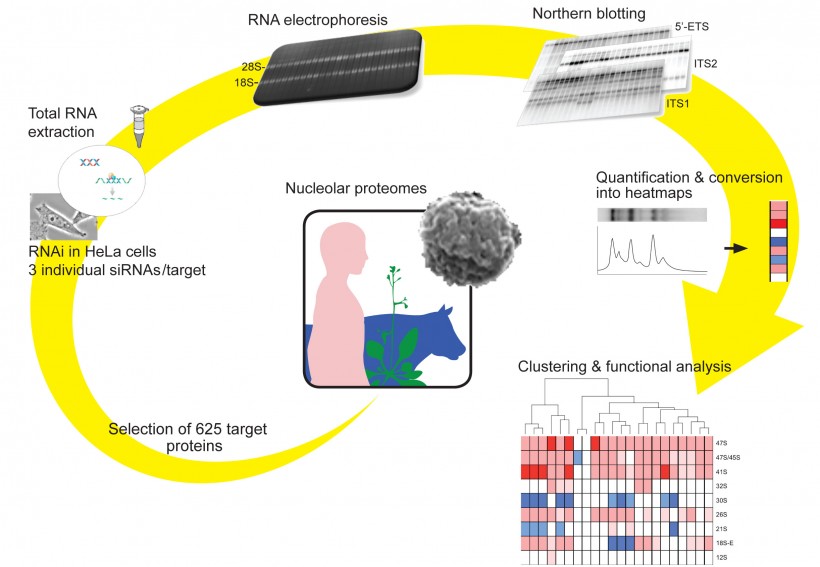
Here, in an effort to better understand ribosome synthesis in human cell lines, we have characterized the function in pre-rRNA processing of a selection of 625 putative nucleolar proteins in HeLa cells. These candidates were selected based on the recently established nucleolar proteomes in human, bovine and plants [2,3,4,5]. The rationale for the screening was twofold. Firstly, many of the initial pre-rRNA cleavages occur within the nucleolus, hence the factors involved should be localized there. Secondly, several years ago, it was established in yeast that many trans-acting factors involved in late assembly steps, i.e. occurring in the nucleoplasm and in the cytoplasm, are loaded onto the pre-ribosomes at an early nucleolar stage [1].
For each gene selected, three silencer RNAs (siRNAs) were tested individually and the depletions were carried out for 3 days. Total RNA was extracted, separated on denaturing agarose gels and analyzed by Northern-blotting. A set of three probes and specific calibration controls were used. The probes used are specific to sequences located in the 5’-ETS, ITS1 and ITS2, and together hybridize to most known pre-rRNA intermediates. Each prominent pre-rRNA species detected, 10 altogether, was quantitated and the data set, representing >2000 individual siRNA depletions, excluding the iterative controls, was assembled in a matrix and integrated with a clustering software. This functional clustering allowed the grouping of the inactivations according to similar patterns of accumulation/reduction of pre-rRNA precursors and, therefore, the designation of specific functional classes.
References
- Henras AK, Soudet J, Gerus M, Lebaron S, Caizergues-Ferrer M, Mougin A, Henry Y (2008) The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol Life Sci 65: 2334-2359.
- Andersen JS, Lyon CE, Fox AH, Leung AK, Lam YW, Steen H, Mann M, Lamond AI (2002) Directed proteomic analysis of the human nucleolus. Curr Biol 12: 1-11.
- Scherl A, Coute Y, Deon C, Calle A, Kindbeiter K, Sanchez JC, Greco A, Hochstrasser D, Diaz JJ (2002) Functional proteomic analysis of human nucleolus. Mol Biol Cell 13: 4100-4109.
- Pendle AF, Clark GP, Boon R, Lewandowska D, Lam YW, Andersen J, Mann M, Lamond AI, Brown JW, Shaw PJ (2005) Proteomic analysis of the Arabidopsis nucleolus suggests novel nucleolar functions. Mol Biol Cell 16: 260-269.
- Patel AK, Olson D, Tikoo SK (2010) Proteomic analysis of bovine nucleolus. Genomics Proteomics Bioinformatics 8: 145-158.